Investigate the mechanism of how Ibuprofen molecules block the aggregation process of Aβ peptide
Introduction
Alzheimer's Disease (AD) pathogenesis is widely believed to be driven by the production and deposition of the amyloid-β peptide (Aβ), which is a ∼ 4 kDa peptide. It has been found that chronic orally administered Ibuprofen, the most commonly used nonsteroidal anti-inflammatory drug, was able to produce significant diminution in the ultimate number and total area of β-amyloid deposits in transgenic mouse model with AD. However, the mechanism is still not fully understood. To investigate the mechanism of how Ibuprofen molecules block the aggregation process of Aβ, designed molecular simulations were performed in this study. The effect of Ibuprofen molecules on the aggregation of Aβ16-22 fragment was studied in the simulation. The secondary structures of Aβ16-22 fragment were measurable outcomes in the simulation to answer the research question.
Materials and Methods
Aβ16-22 fragment structure was obtained from PDB, solvated either in the water system or water+Ibuprofen system. Simulation was completed in four steps, minimization, heating, equilibration and quenching. The consistency of each simulation was ensured by examining the corresponding output file. The secondary structure of each residue was obtained from quenching output files, which were based on the coordinates of each atom and identified by STRIDE in VMD.
Results
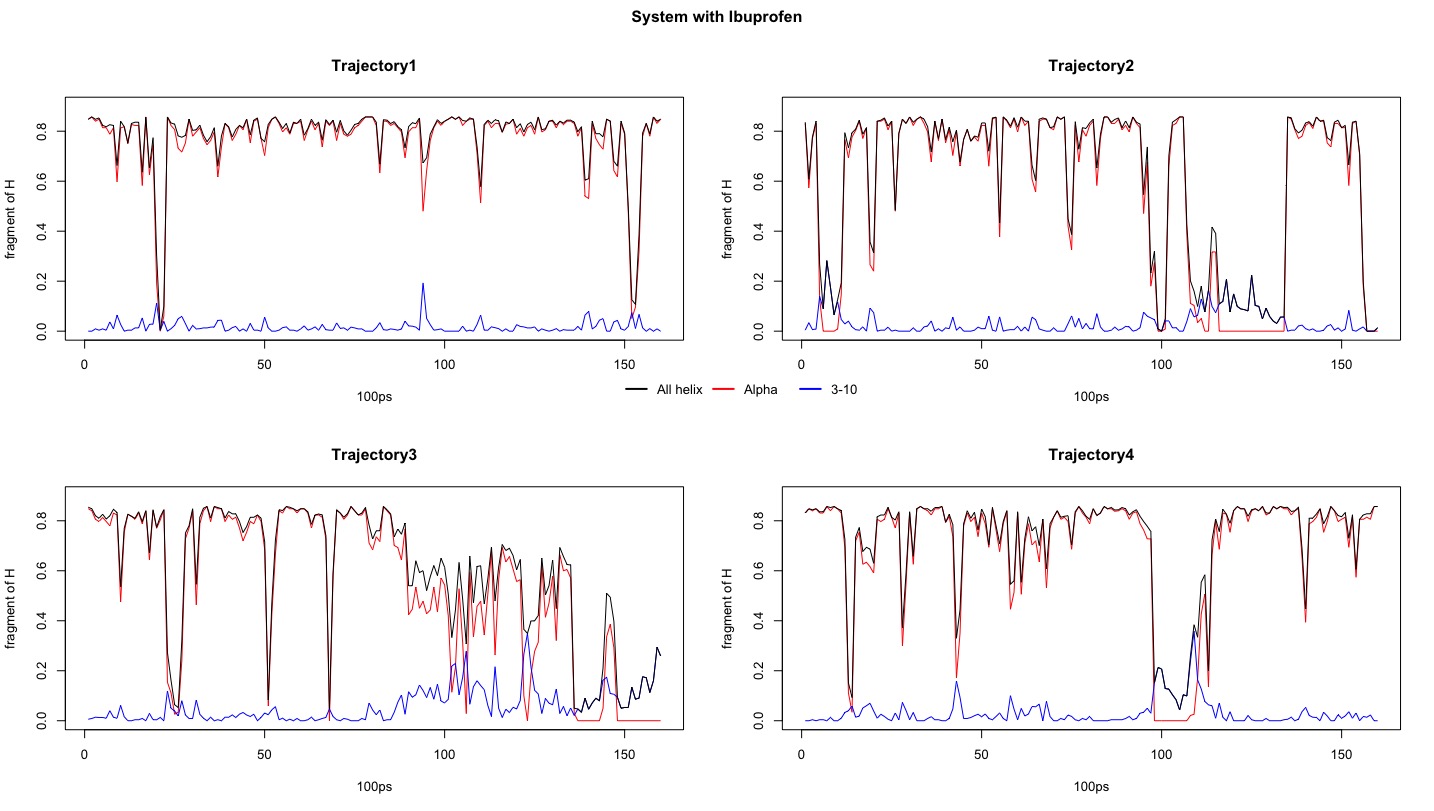
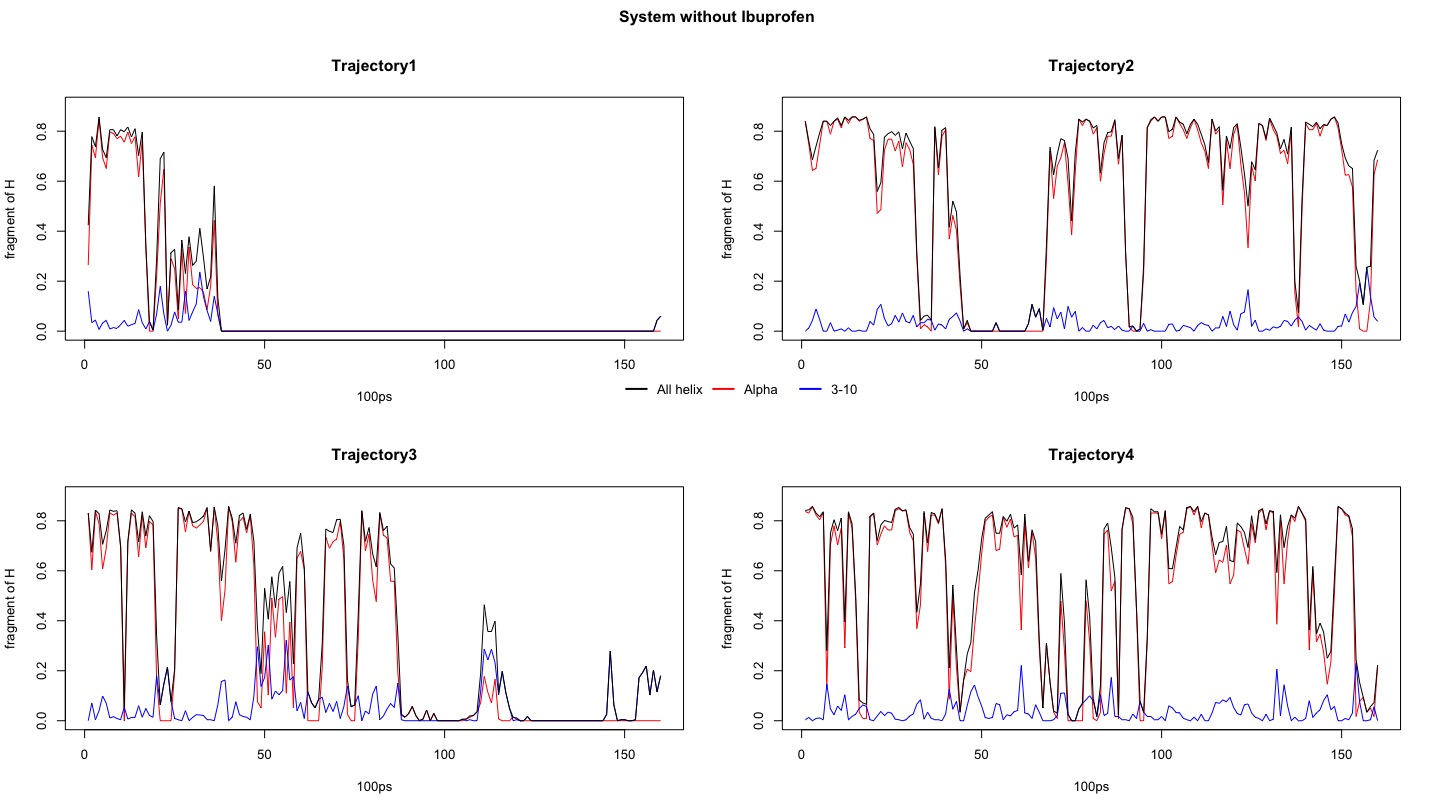
Figure1. Fragment of all helix, alpha helix and 3-10 helix within system in different trajectories
The number of secondary structures of 310-helix was also obtained. It reached peak of approximate 0.15 at residue 3 and 4 in Trajectory 3 in Experimental system, while the peak was also approximate 0.15 but at residue 2, 3 and 4 in Trajectory 3 in control system. Therefore, there was no difference in fraction of 310-helix secondary structure between Experimental and Control systems.
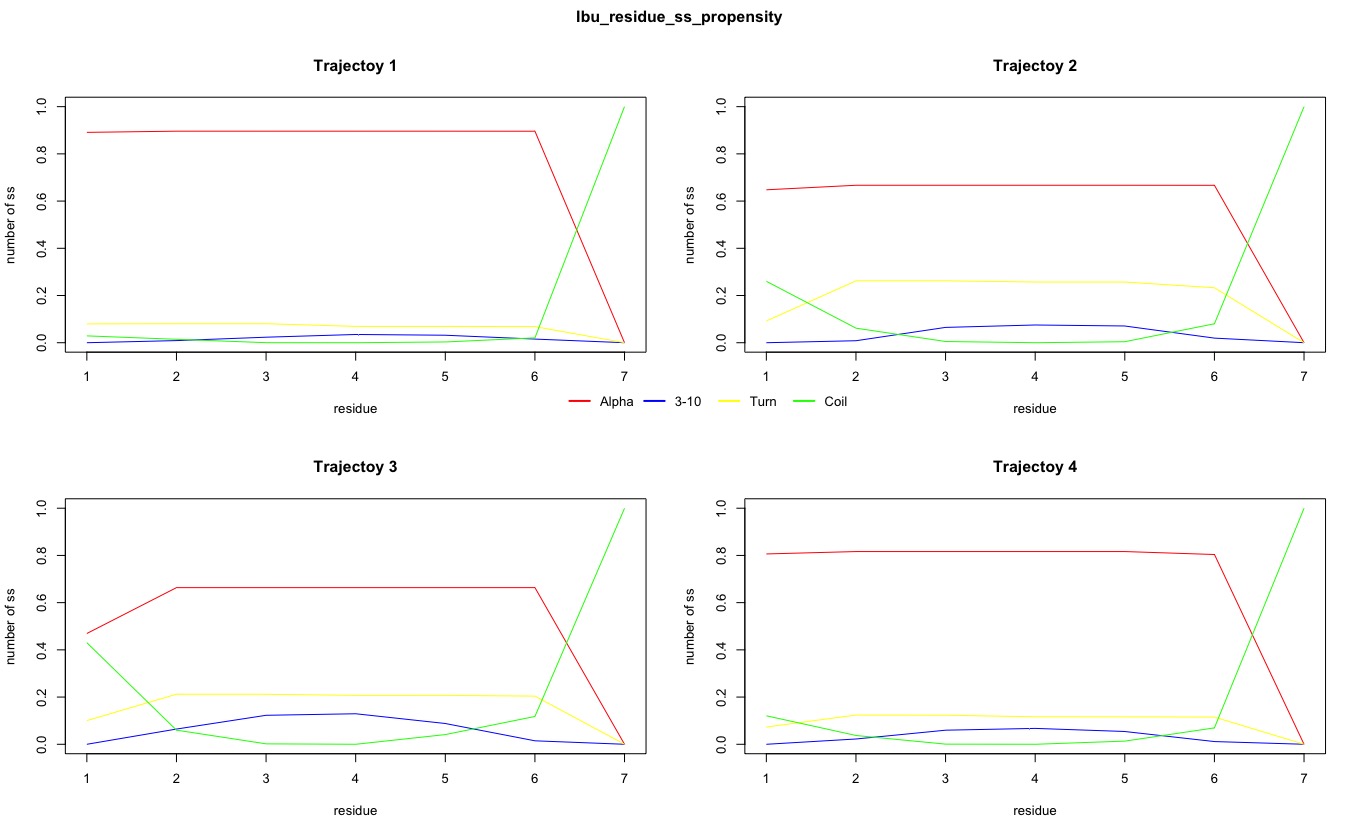
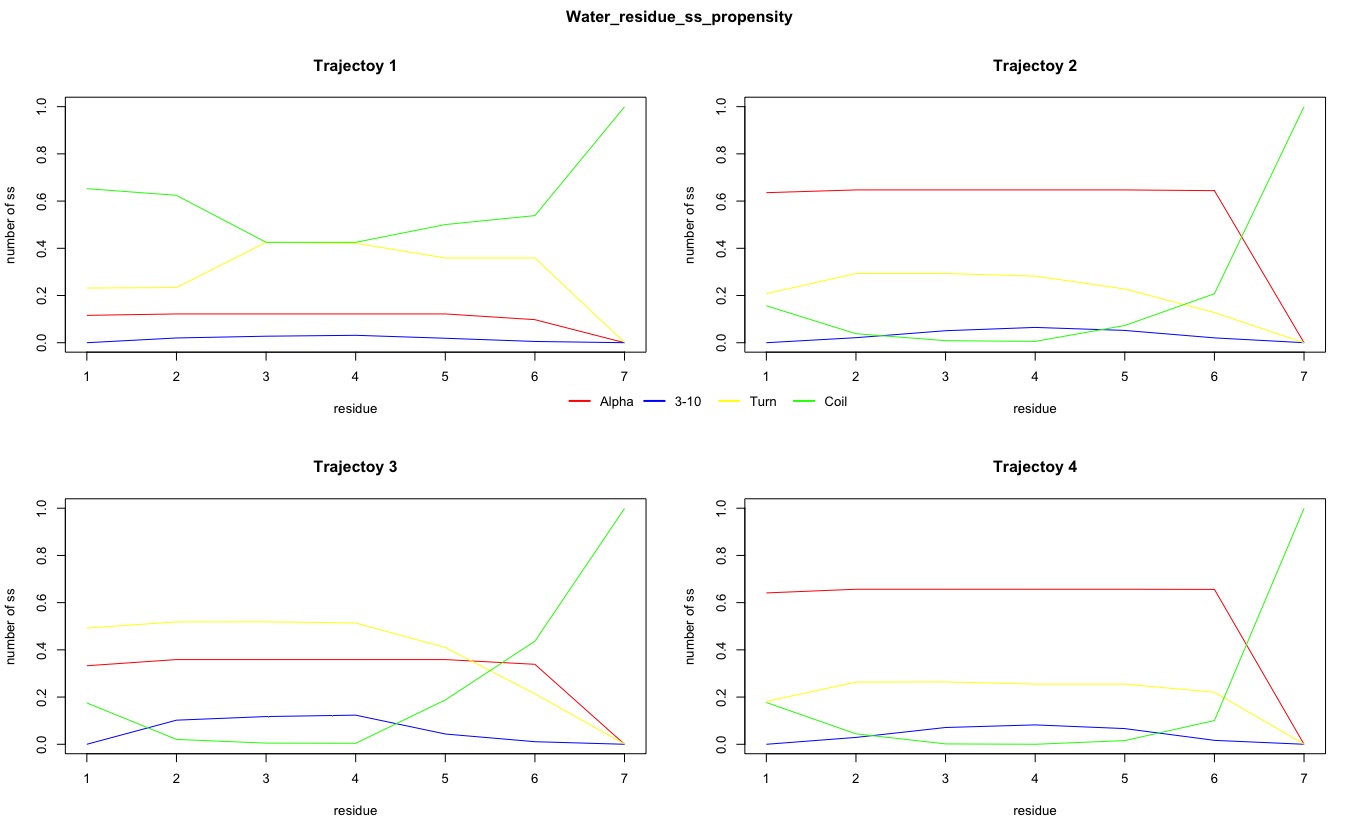
Figure2. Propensities of different secondary structure for each residue