|
AUTO–MUTE |
|
|
AUTO-MUTE Home |
Disease Potential of Human nsSNPs DocumentationIntroductionDisease Potential of Human
nsSNPs is an
automated server for predicting whether a human non-synonymous single
nucleotide polymorphism (nsSNP) in a coding region, causing a single
amino acid replacement in the corresponding protein structure, has an
association with disease or is simply neutral. The approach is based
upon the intuitive notion that the effects of nsSNPs, either benign or
leading to aberrant protein function and disease, correlate well
with relative structural changes from wild-type. A dataset
of 1790 single amino acid substitutions,
each corresponding to either a neutral (nt) or a disease-associated
(da) nsSNP, was used for model training and validation. The variants
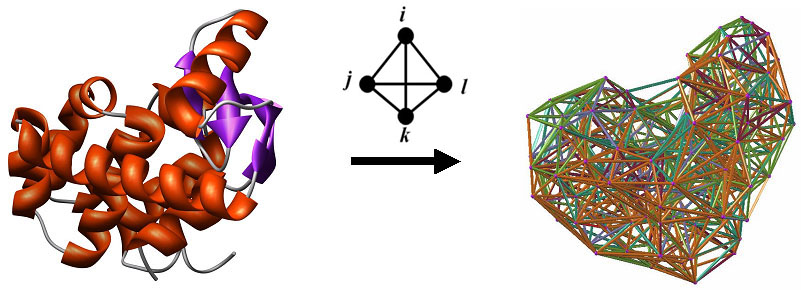
map to positions within 243 tessellatable, single chain, wild-type
protein structures in the Protein
Data Bank (PDB). In particular, the dataset consists of 458 ntSNPs
mapping to 184 structures and 1332 daSNPs mapping to 102 structures.
Our server consists of a
supervised classification
model based on implementation of the Random Forest (RF) algorithm. In
addition to these nsSNPs with known functional effects, we also
collected 357 unclassified (unSNPs) human nsSNPs mapping to 44
tessellatable, single chain, wild-type
protein structures, and we classified each of them (as either nt or da)
based on predictions obtained from our RF model (see Results section below).
The set of human nsSNPs described above were selected from among a significantly larger collection of human single amino acid variants appearing in the Swiss-Prot database (release 51.3, 12-Dec-2006), whose accession files contained PDB cross-references to x-ray crystallographic protein structures. Furthermore, we only considered a variant if it occurred at a Swiss-Prot sequence position mapping to a position within a tessellatable, single chain in at least one of the PDB cross-reference structure files, and the position undergoing mutation was also required to have at least six tessellation-based nearest neighbors. The resulting dataset of selected nsSNPs provides Swiss-Prot and PDB identification information for each variant, as well as the class (da, nt, un). Lastly, a server prediction about a human nsSNP can only be performed if the variant represents a single amino acid substitution at a position within a tessellatable, single chain of a solved protein structure with a coordinate file available in the PDB, and the position to be mutated must have at least six tessellation-based nearest neighbors. Specifically, Delaunay tessellation of the structure can be performed if the PDB file contains consecutive primary sequence numbering in the ATOM lines (i.e., no gaps in the structure) starting with a non-negative integer, the alpha-carbon atomic coordinates are available for all the constituent amino acids, and no alternative conformations exist for the alpha-carbon atoms. In addition to X-ray structures, NMR structure files are potentially tessellatable if they consist of a single minimized average structure as opposed to multiple models. Methods Among the numerous factors influencing model performance are the dataset size and composition utilized for training, the type of attributes (i.e., predictors) used as components for the feature vectors characterizing the variants in the dataset, and the machine learning algorithm chosen for model building. AUTO-MUTE utilizes attributes that include EC score at the mutated position (mutant protein residual score), ordered EC scores of the six nearest neighbors to the mutated position, native and replacement amino acid identities at the mutated position, ordered amino acid identities at the six nearest neighbors, and ordered differences between the primary sequence positions of the nearest neighbors and the mutated residue (see AUTO-MUTE home page for details). Additionally, the following Delaunay tessellation-derived attributes were utilized as feature vector components for each variant: mean volume and tetrahedrality of the simplices in which the mutated position serves as a vertex, location (surface, undersurface, or buried) of the mutated position, number of edge contacts that the mutated position has with surface positions, and secondary structure (helix, strand, coil, or turn) of the mutated position (see AUTO-MUTE home page for details). The dataset of feature vector components for each of the selected nsSNPs provides the raw data used for training the RF model and predicting the unSNPs, and the PDB accession code, chain, and variant PDB position number are also provided only as a means to identify each mutant. Required Inputs and Server Outputs A valid PDB accession code and a specific chain (use @ if null) is required for the structure of the human protein containing a single residue substitution whose functional impact (neutral or disease-associated) is to be predicted. The variant under consideration must be supplied in the form (native residue)(position number from PDB file ATOM lines)(replacement residue), for example I53T. In addition to reproducing the inputs, the output data includes the prediction (neutral or disease-associated) along with a confidence measure, mean volume and tetrahedrality for the mutated position, location and number of edge contacts that the mutated position has with surface positions, and secondary structure of the mutated position. Results We utilize both 20-fold and leave-one-out (LOO) cross-validation (CV) procedures on the dataset of 1790 neutral and disease-associated nsSNPs, and we apply a random split of the dataset (66% for RF model training, 34% for testing). Performance is evaluated by calculating the following values, where TP (TN) = total number of correctly predicted “disease-associated” (“neutral”) mutants, and FN (FP) = total number of respectively misclassified mutants. The overall accuracy, while not ideal in the case of significant class skew, is defined as Q = (TP + TN) / (TP
+ TN + FP + FN).
Hence, the following measures are also calculated due to their robustness with respect to unequal class distributions: balanced error rate is defined as BER = 0.5 ×
[FN / (FN + TP) + FP / (FP + TN)],
Matthew’s correlation coefficient is given by MCC
= (TP ×
TN – FP
× FN) / [(TP + FN)(TP
+ FP)(TN + FN)(TN + FP)]1/2,
and AUC refers to
area under the receiver operating characteristic (ROC) curve, a plot of
true positive rate (i.e., TP / (TP + FN), or sensitivity) versus false positive
rate (i.e., FP / (FP + TN), or 1 – specificity)
for the disease-associated class (defined analogously for the neutral
class).
*average over ten
independent iterations
By using the best 20-fold CV iteration above, as well as performing a 10-fold CV procedure, we compare our results with those of other methods as summarized below. Keep in mind that an absolutely direct comparison is not possible here because of differences in algorithms (e.g., RF versus SVM), training datasets (ours requires variant positions within tessellatable structure cross-references and is significantly smaller than those used by the other methods), and feature vector components (our attributes are novel tessellation-based values that are distinct from those used by the other methods). Given these considerations, the performance results below suggest that the signals embedded in our feature vectors are either as informative, or significantly more so, for accurate class discrimination.
____________________________________________________________________________________
The trained Auto-Mute RF model was subsequently used for generating unSNP class predictions (either nt or da) for each of the 357 unclassified variants obtained from Swiss-Prot. References
|