|
AUTO–MUTE |
|
|
AUTO-MUTE Predictors: ========================== Alternative Protein-Specific
Models
Mutant Activity Databases
Structural
Bioinformatics at or
|
**NEW!**
Portable AUTO-MUTE 2.0 with
enhanced features (batch jobs,
non-PDB models, multiple model NMR
files, etc.), a command-line
driven alternative: free,
platform-specific downloads available for PC (AutoMute2.zip), Mac (AutoMute2.tar.gz), and Cygwin (cygwin_AutoMute2.tar.gz). All versions require Perl (with CPAN library), Java, and internet access (for automatic PDB file downloads); Cygwin users should first download and install Qhull from the online package list. The unzipped AutoMute2 folder contains the file weka.jar, to which the computer's CLASSPATH environment variable must point (requires advanced computer skills). Alternatively, make the following simple edits to five Perl scripts (three programs named stability_changes_XX.pl, as well as activity_changes.pl and human_nsSNPs.pl) in the unzipped directory after opening them in any text editor (e.g., Wordpad). Within each of the stability_changes_XX.pl files, scroll midway to find four lines starting with "open(RESULT, 'java ...", and change them to "open(RESULT, 'java -cp weka.jar ...". For the remaining two Perl programs, each has only one such line that requires identical editing. WELCOME TO THE AUTO-MUTE SUITE OF PREDICTORS... ... harnessing the
combined power of computational mutagenesis using a four body,
knowledge-based potential, along with
cutting-edge machine learning methodologies and tools, to
provide more accurate predictive models of mutant protein function.
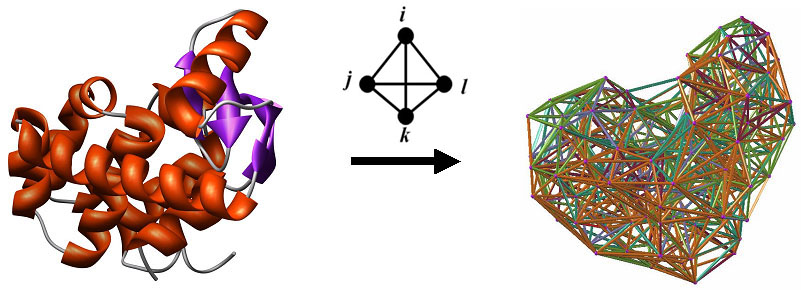
For each type of function prediction, a variety of classification and regression models have been developed and are available for researchers. These include Random Forest, Support Vector Machine (SVM), AdaBoostM1 combined with the C4.5 Decision Tree algorithm, as well as Tree and SVM regression. Details concerning the datasets used for training and the performance of these models is available in the form of additional documentation linked to the respective server pages, and publications will be forthcoming. First, protein structures are reduced to collections of points in 3-dimensional space, whose coordinates are those of amino acid alpha-carbon atoms. Next we apply Delaunay tessellation to each discretized protein structure, whereby the points are utilized as vertices for tetrahedral simplices that tile the space and identify quadruplets of nearest-neighbor amino acids in each protein. To safeguard against quadruplets that do not interact biologically, only tetrahedra whose six edges are all less than 12 Angstroms are considered. The approach is applied to a training set of over 1400 high-resolution x-ray structures with low sequence and structure similarity obtained from the Protein Data Bank (PDB), and normalized frequencies of occurrence (fijkl) are calculated for each of the 8855 order-independent quadruplets possible from the 20 naturally occurring amino acids. The multinomial distribution (n = 4) is used to also compute an expected rate of occurrence (pijkl) for each quadruplet type. A log-likelihood score (potential), given by qijkl = log (fijkl/pijkl), measures propensity of occurrence for each quadruplet type. A residue environment score is calculated for every amino acid in a protein by summing the log-likelihood scores of all simplex quadruplets in which it participates as a vertex, yielding a 3D-1D potential profile. By utilizing the tessellation of the wild type protein structure, a novel computational mutagenesis is defined by changing the residue label at a position of interest and re-computing the environment scores. Only the mutated residue position, as well as all neighboring positions that participate as vertices in simplices with the mutated position, will experience alterations in their environment scores in the mutant 3D-1D potential profile relative to the wild type profile. We refer to the vector difference of these profiles (mutant - wild type) as the residual profile of the mutant protein, and its components as EC (environmental change) scores that quantify perturbations at the corresponding residue positions in the protein. A common set of attributes for single residue substitutions across all protein structures is defined as follows. First, we consider the EC score of the mutated position, also referred to as the residual score of the mutant protein. Next, we include the EC scores of the six nearest-neighbor positions that participate in simplices with the mutated position, ordered by simplex edge-length Euclidean distance away from the mutated position. Finally, we also utilize the wild type and replacement amino acid identities, the ordered identities of the amino acids at the six nearest neighbors, and the ordered differences between the primary sequence positions of the nearest neighbors and the mutated residue. The ordering of the latter two sets of attributes for the six nearest neighbors is identical to that of their EC scores. Instead of including relative solvent accessibility (RSA) of the mutated position as an attribute, we compute the following tessellation-based quantities yielding models which perform equally well. A mean volume and tetrahedrality is calculated for all simplices in which the mutated position serves as a vertex. If a mutated position participates as a vertex in a triangular face of a simplex, and if that triangular face is not shared by another simplex, then the position is referred to as being on the Surface. If a mutated position does not satisfy this property, but at least one simplex edge connects the mutated position to a Surface position, then the mutated position is referred to as Undersurface. All other positions are considered Buried. A count is obtained for the number of edge contacts that the mutated position has with surface positions; buried mutated positions have a count of zero by definition. Secondary structure of the mutated position is also included as an attribute (helix, strand, coil, or turn). Lastly, for certain predictors we include temperature and pH of experimental conditions under which measurements (ΔΔG, ΔΔGH2O) were collected for the mutant proteins. References
|